Macaca fascicularis and Macaca mulatta are the most commonly used non-human primates in experimental research and are widely utilized in biomedical and human evolutionary studies due to their close genetic affinity to humans (having diverged around 25 million years ago), particularly in the fields of human trait formation, disease modeling, and drug metabolism. Their reference genome has been widely applied in these studies, especially in understanding human characteristic trait formation, disease modeling, and drug metabolism. However, the existing reference genomes still contain many unknown sequences, particularly in the regions of the centromere, segmental duplications (SDs), and ribosomal DNA (rDNA), which limits the in-depth exploration of primate evolutionary mechanisms and their biomedical value.

Figure 1 Screenshot of the first page of the paper published in Nature
On February 26, 2025, Yafei Mao’s group at the Bio-X Research Institute of SJTU, in collaboration with Qiang Sun’s group at the Center for Excellence in Brain Science and Intelligence Technology/Institute of Neuroscience of the Chinese Academy of Sciences (CAS), published a paper in the journal Nature entitled Integrated Analysis of the Complete Sequence of a Macaque Genome (Figure 1). In this study, the complete telomere-to-telomere (T2T) genome of a non-human primate was assembled for the first time, and the large-scale genomic differences between macaques and humans were systematically analyzed. The study also explored how structural variations regulate type-specific expression of brain cells through three-dimensional genome reconstruction, using the FOLH1 gene family as a key case. Additionally, the study revealed the genetic characteristics of interspecies differentiation within the macaque genus, providing a crucial genetic foundation for the biomedical modeling of non-human primates.
1. Filling gaps in complex regions
Conventional genome assembly techniques have a large number of genetic gaps due to short read lengths and high error rates, which make it difficult to span complex repetitive regions. These gaps are mostly located in the complex structural regions of the genome (such as centromere, SD, and rDNA), which are precisely the important parts of the genome structure and function, and are involved in biological processes such as chromosome stability, gene expression regulation, and chromosome rearrangement.
In order to solve this challenge, the research team first constructed a solitary female reproductive embryonic stem cell line (MFA582-1) in Macaca fascicularis [1]. The solitary female reproductive cell line has nearly homozygous genetic information of two sets of chromosomes, this characteristic makes it an ideal material for constructing a reference genome. On this basis, the team utilized the self-developed local assembly tool based on the unique k-mer markers for iterative substitution of typing, and successfully solved hundreds of complex structural regions that were not assembled or misassembled by the existing assembly software, and finally constructed the T2T genome of the Macaca fascicularis T2T-MFA8v1.1. The genome has a total length of 3.06 Gbp, with a QV of 71.27 and an NG50 of 162.13 Mbp, which is comparable to that of the human genome T2T-CHM13v2.0, making it the first complete reference genome for non-human primates, and providing an important tool for in-depth understanding of complex genomic regions. The research team found:
Specificity of SD distribution patterns: the total length of SDs in Macaca fascicularis, 122.51 Mbp, is 46% less than that of humans (227.36 Mbp) and is more enriched in the subtelomeric region (humans are biased toward the proximal centromere region). This distributional difference may drive the directionality of primate genome remodeling.
rDNA distribution: rDNA is localized only on chromosome 10 in the Macaca fascicularis, whereas in the human genome, rDNA is distributed on five proximal centromere chromosomes (chr13, chr14,chr15, chr21, chr22). This difference provides a new explanation for the formation of proximal centromere chromosomes in humans and the evolutionary medical mechanism of the human disease trisomy 21.
Structural evolution of centromere: Macaca fascicularis centromeres are dominated by α-satellites of the SF7 family, which are on average 3.83-fold longer than those of humans (e.g., chr15 reaches 13.88 Mbp) and retain the ancient SF8-SF13 sequence layer that was coexisting with the primate ancestor, which reveals the existence of a distinctive evolutionary mechanism for the formation of new centromere in the genus Macaca.
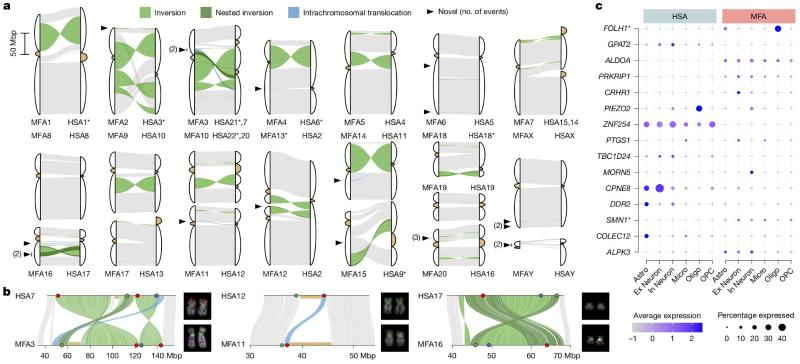
Fig. 2 Fixed large-scale structural variation in human and genus Macaca
2. Genomic differences due to large-scale structural variation in human and genus Macaca
Large-scale structural variations (e.g., inversions and translocations) in the genome can alter the three-dimensional folding of chromatin, which in turn affects gene expression patterns. However, large-scale structural variations in the genomes of humans and genus Macaca have not yet been fully analyzed, and there is still a lack of systematic evidence to support the functional effects of these structural variations in primate evolution.
To solve this problem, the research team optimized the identification of structural variants, and identified 93 fixed structural variants between human and genus Macaca, including 78 inversions, 11 Centromere relocations, and 4 intrachromosomal translocations, of which 21 structural variants were reported for the first time (Figure 2). Further studies revealed that more than 400 genes may undergo differential expression in brain cell taxa due to related structural variants during primate brain evolution. Taking the FOLH1 gene as an example, its encoding glutamate carboxypeptidase II (GCPII) plays a key role in glutamate regulation in the nervous system, and mutations in the gene are closely associated with mental retardation [2]. In the genus Macaca, the FOLH1 gene is a single copy, whereas in humans, it contains two copies, FOLH1 and FOLH1B (directly homologous to macaque FOLH1). The human FOLH1 gene is highly expressed in oligodendrocytes, whereas the FOLH1B gene is barely expressed in different cellular taxa of the brain, which is significantly different from the pattern of widespread expression of FOLH1 in multiple cellular taxa in the macaque genus.
In order to reveal the cause of this difference, the team integrated single-cell multi-omics data and found that a 1.4 kbp fragment of the human FOLH1B regulatory region was specifically lost during the evolution of humans and macaque genus, leading to its “pseudogenization” by not being expressed in the brain. At the same time, the human FOLH1 gene underwent a repetitive event that led to the remodeling of the three-dimensional structure of chromatin, which in turn altered its cellular taxon expression pattern (Figure 3). This study provides new insights into how structural variation affects cell-type-specific expression patterns during evolution, and is particularly important for elucidating the formation of lineage-specific phenotypes and the mechanisms of human diseases.
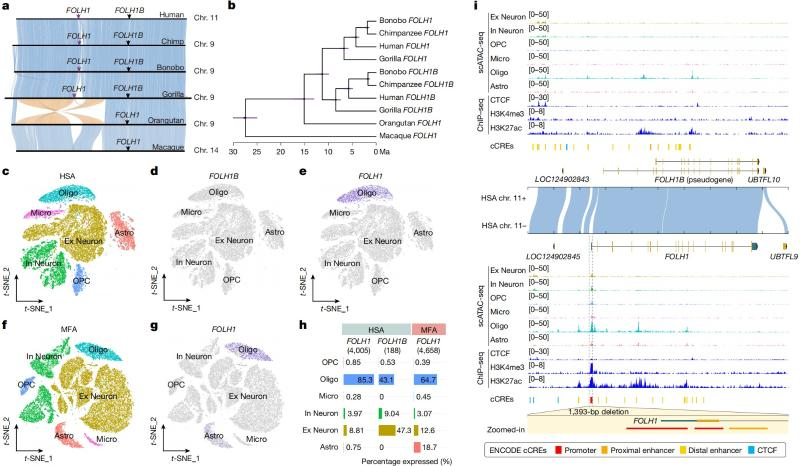
Figure 3 Evolutionary history and multi-omics analysis of the FOLH1 gene family
3. Interspecific genetic differentiation from sequence to phenotype between Macaca fascicularis and Macaca mulatta.
Macaca fascicularis and Macaca mulatta belong to the same genus Macaca, but there are significant differences in morphological characteristics, behavioral patterns and disease susceptibility. However, the genetic basis of these phenotypic differences has not been fully elucidated, thus severely limiting the potential application of these two species in biomedical modeling.
By means of pan-genome mapping and other means, the research team identified 240 Mbp regions of complex structural differences between species, covering gene families such as Mafa-AG/B, CYP2C76 and GSTM. Among them, Macaca mulatta CYP2C76 (monkey-specific cytochrome P450 enzyme gene) showed four structural haplotypes, while Macaca fascicularis retained only two, which may reflect the metabolic differences between the two species (Figure 4). In addition, the team identified 16.76 Mbp of genetically differentiation regions (Figure 4) between Macaca fascicularis and Macaca mulatta, and found that the fixed genetic differences between the two species appeared to be 9.43-fold enriched in regulatory elements, including the HOXD13 gene, which may affect the length of the tail. This finding coincides with the phenotypic characterization of Macaca fascicularis with significantly longer tails than Macaca mulatta. In addition, this finding also forms a case of convergent evolution with the study of adaptive evolution of the deer mouse tail [3], which provides a new perspective to unravel the genetic mechanisms underlying the morphological diversity of mammalian tails.
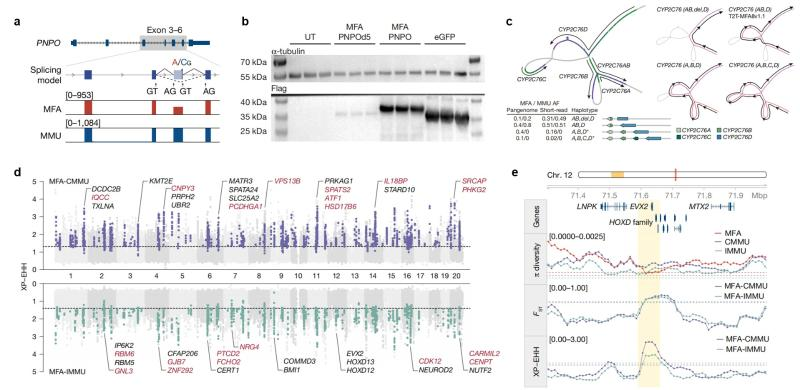
Fig. 4 Interspecific genetic differentiation between Macaca fascicularis and Macaca mulatta
Overall, this study systematically elucidates the evolutionary differences between the macaque genus and humans at the genome structure level by developing a novel computational tool to achieve the complete telomere-to-telomere (T2T) genome assembly of a non-human primate. It not only reveals the cell-type specificity of structural variations affecting gene expression through modulation of three-dimensional genome remodeling and alterations of regulatory elements, but also deeply analyzes the genetic basis of interspecific differentiation within the macaque genus, laying a solid genetic foundation for biomedical modeling of macaques. This study enhances our understanding of primate evolutionary medicine, biomedical modeling, and lineage-specific adaptation.
This work is representative of a series of research results by Yafei Mao’s team supported by the Bio-X Research Institute of SJTU. Academician Lin He of the Bio-X Research Institute of SJTU pointed out that “this achievement is a classic case of large-scale chromosome variation research, and the related work provides a new perspective for analyzing the biomedical functions of complex structural variation in primate genomes, promotes the cross development of informatics, evolutionary biology and medical genetics, and lays an important foundation for future research in the field of evolutionary medicine”.
Yafei Mao from the Bio-X Research Institute of SJTU and Qiang Sun from the Center for Excellence in Brain Science and Intelligence Technology/Institute of Neuroscience of the CAS are the co-corresponding authors of the paper. This work was greatly supported by experts and scholars from SJTU, the Center for Excellence in Brain Science and Intelligence Technology of the CAS, Zhejiang University, the Kunming Institute of Zoology of the CAS, and the Chengdu Institute of Biological Sciences of the CAS. Shilong Zhang, Ning Xu and Lianting Fu of SJTU and the Center for Excellence in Brain Science and Intelligence Technology of the CAS are the co-first authors of the paper. This study was conducted in collaboration with several primate consortia at home and abroad, and we would like to appreciate the consortia for their data sharing support.
Yafei Mao’s research group at the Bio-X Research Institute of SJTU focuses on primate evolutionary medicine, using interdisciplinary approaches such as evolutionary biology, computational biology, neurobiology, and large-scale functional screening to analyze the genetic mechanisms underlying the formation of primate-specific adaptive traits and the occurrence of human disease risk loci. The research group is recruiting postgraduate students, postdoctoral fellows, and research assistants with backgrounds in evolutionary biology, bioinformatics, cell biology, neuroscience, computational science, and related fields, offering a broad development platform for talented individuals interested in primate evolutionary medicine research. If you are interested in the group's research direction and have the relevant professional background and skills, you are welcome to visit the research group’s website (https://www.yafmao.org/) for more information.
Paper link:
https://www.nature.com/articles/s41586-025-08596-w
References:
1. Yang, H. et al. Generation of haploid embryonic stem cells from Macaca fascicularis monkey parthenotes. Cell Research 23 (2013). https://doi.org/10.1038/cr.2013.93
2. Rahn, K. A. et al. Inhibition of Glutamate Carboxypeptidase II (GCPII) activity as a treatment for cognitive impairment in multiple sclerosis. Proceedings of the National Academy of Sciences 109 (2012). https://doi.org/10.1073/pnas.1209934109
3. Kingsley, E. P. et al. Adaptive tail-length evolution in deer mice is associated with differential Hoxd13 expression in early development. Nature Ecology & Evolution (2024).


